
Informations générales
Événement : 90e Congrès de l'Acfas
Type : Colloque
Section : Section 100 - Sciences de la santé
Description :La biologie structurale est un domaine de la recherche en biologie qui étudie l’organisation et le fonctionnement des mécanismes biologiques et de ses constituants, à l’échelle atomique. Ces mécanismes incluent autant ceux impliqués dans les fondements du vivant (transcription et réparation de l’ADN, traduction génétique, respiration cellulaire, autophagie) que ceux impliqués dans diverses pathologies (maladies infectieuses, cancers, vieillissement, dérèglements génétiques). Il est remarquable que les mécanismes biologiques reposent sur les fonctions d’un nombre limité de types de macromolécules naturelles, principalement les protéines, les acides nucléiques, les sucres et les lipides, et sur leurs interactions. Ainsi, la biologie structurale s’intéresse à examiner en trois dimensions à l’échelle atomique la complexité et la diversité de ces différents types de macromolécules biologiques et leur fonctionnement.
Ce volet des sciences fondamentales en médecine utilise une grande variété de techniques biophysiques pour visualiser les molécules du vivant et mieux comprendre leur fonctionnement, telles la résonance magnétique nucléaire (RMN), la cristallographie aux rayons X, la diffusion de rayons X aux petits angles (SAXS) et la cryo-microscopie électronique (cryo-EM). Grâce à cette instrumentation de pointe, la recherche en biologie structurale permet de faire des découvertes importantes sur la base atomique et moléculaire des phénomènes biologiques et de faire progresser les connaissances sur le rôle des macromolécules biologiques dans les nombreuses pathologies infligeant le vivant.
Notre colloque sert de vitrine pour mettre de l’avant les récentes découvertes en ce sens où des scientifiques d’ici et d’ailleurs ayant démarré leur programme de recherche indépendant dans les dernières années présentent leurs recherches. Des présentations orales et par affiches offertes par des étudiantes et étudiants aux cycles supérieurs ainsi que par des stagiaires postdoctoraux complètent le programme.
Date :Format : Sur place et en ligne
Responsables :- Normand Cyr (UdeM - Université de Montréal)
- Malik Chaker-Margot (UdeM - Université de Montréal)
- Pascale Legault (UdeM - Université de Montréal)
Programme
Accueil, installation des affiches et mot de bienvenue
Accueil des participants et participantes, installation des affiches et mot de bienvenue.
Signalisation et cancer
-
Communication orale
Mécanismes moléculaires de la détection de stress mitochondrial par PINK1Jean-François Trempe (Université McGill)
Des mutations dans la protéine PINK1 provoquent une forme familiale de la maladie de Parkinson (MP), une maladie neurodégénérative caractérisée par des troubles du mouvement. PINK1 est une kinase avec une séquence de localisation mitochondriale qui est coupée par des protéases dans la membrane interne et la matrice, avant d’être exportée vers le cytosol pour être dégradée. Comme le taux de dégradation excède le taux de traduction, on retrouve peu de protéine PINK1 à l’état normal. PINK1 agit comme un détecteur de stress mitochondrial en s’accumulant sélectivement sur les organelles endommagées, où elle forme un complexe avec la translocase de la membrane externe (TOM). L’ancrage de PINK1 au complexe TOM permet son autophosphorylation suivie de la phosphorylation de l’ubiquitine, un prérequis pour la mitophagie. Nous avons déterminé la structure du domaine kinase de PINK1 par cristallographie à rayons X à une résolution de 2.8 Å. Le protéine forme un dimère qui explique comment PINK1 s’autophosphoryle en trans sur la sérine 205, une étape cruciale pour son activation. Finalement, nous démontrons que les extensions N- et C-terminales, qui contiennent des mutations associées à la MP, forment un module d’interaction avec TOM. Ces résultats mènent à une meilleure compréhension du mécanisme d’activation de PINK1 sur les mitochondries.
-
Communication orale
Structure et signalisation des récepteurs aux prostaglandinesMartin Audet (UdeS - Université de Sherbrooke)
Les prostaglandines sont des médiateurs lipidiques impliqués dans la régulation de plusieurs conditions physiologiques tels que l’asthme, la pression artérielle, les maladies cardiovasculaires, les accouchements prématurés et les cancers. Ils agissent sur des récepteurs couplés aux protéines G pour activer des cascades signalétiques intracellulaires qui dictent leurs réponses physiologiques. Le récepteur à la prostaglandine E2 de type 3 (EP3) est ciblé par la prostaglandine E2 (PGE2) et régule les fonctions rénales, la fièvre, les contractions du myomètre, mais aussi les systèmes immunitaires et vasculaires. Il constitue une cible émergente pour le traitement du diabète de type 2, de l’asthme, et de l’athérosclérose. Au moins 12 isoformes du récepteur EP3 ont été recensées et elles ne diffèrent que par la séquence du domaine c-terminale. La compréhension structurale du mode de liaison et de signalisation des prostaglandines sur le récepteur EP3 est la clef pour le développement des nouvelles thérapies ciblant le récepteur. Dans cette présentation, la structure à haute résolution du récepteur EP3 incluant une analyse du mode d’action des prostaglandines, et les évènements signalétiques engagés par les prostaglandines pour chaque isoformes seront discutés. Ces données donnent une meilleure compréhension de la relation structure-fonction du récepteur EP3 qui guidera le développement de nouvelles molécules ciblant ce récepteur.
-
Communication orale
Mécanismes moléculaires de la regulation de l'activation de la petite GTPase RacMalik Chaker-Margot (UdeM - Université de Montréal)
La sous-famille de GTPase Rac régule le remodelage du cytosquelette d'actine et promeut la prolifération cellulaire. Pour cette raison, l'activation de Rac est associée avec le développement du cancer et des métastases. Comme d'autres petites GTPases, l'activation de Rac nécessite des protéines auxiliaires qui agissent comme facteurs d'échange de guanosine (GEF). Parmi celles-ci, on compte notamment Prex et Tiam, de large protéines multi-domaines qui activent Rac via leur domain catalytique DH-PH. La régulation de l'activité de Prex et Tiam et notamment gouvernée par des interactions protéines-protéines avec des facteurs de signalisation. En utilisant la cryo-microscopie électronique et des techniques biophysiques, nous déterminons le rôle de ces interactions dans la régulation de Prex et Tiam, et ainsi l'activation de Rac1 et du remodelage du cytosquelette.
Microbiologie
Présentation par notre conférencière invitée.
-
Communication orale
Étude de mécanismes spécialisés d'initiation de la traduction chez les parasitesMélissa Léger-Abraham (Harvard Medical School)
L’initiation de la traduction est hautement régulée et requiert la participation d’une quinzaine de facteurs. Le facteur d’initiation eucaryote (eIF) 4E lie la cap des ARNm, (m7GpppX, où G est une guanosine et X est un nucléotide quelconque) localisée à l’extrémité 5’ des ARN messagers (ARNm). eIF4E lie aussi le facteur d’initiation 4G (eIF4G). En retour, eIF4G interagit avec le facteur eIF3, permettant le recrutement de la petite sous-unité ribosomique à l’extrémité 5’ des ARNm, et par conséquent, la formation du complexe de pré-initiation (PIC). Au cours des dernières années, la structure de divers facteurs d’initiation, tel qu’eIF4E, a été obtenue par résonance magnétique nucléaire (RMN) et par cristallographie aux rayons-X. De plus, plusieurs structures décrivant diverses conformations du ribosome au cours de la traduction ont été déterminées par microscopie électronique cryogénique et par cristallographie aux rayons-X. Par conséquent, les mécanismes moléculaires décrivant l’initiation de la traduction chez les cellules de mammifères sont généralement bien compris. Toutefois, ce n’est pas le cas chez les parasites. Ce manque de connaissances limite le potentiel d’interventions thérapeutiques ciblant le processus traductionnel chez les parasites, tels ceux causant, entre autres, la Leishmaniose et Malaria.
Lunch
Boîtes à lunch servies aux participants et participantes du colloque.
Microbiologie et développement de méthodes
-
Communication orale
La RMN de l’état solide pour étudier le mécanisme d’action d’antibactériens in celluloIsabelle Marcotte (UQAM - Université du Québec à Montréal)
Le problème mondial de la résistance des bactéries aux antibiotiques courants stimule la recherche d’approches thérapeutiques impliquant de nouveaux mécanismes d’action. Les molécules qui ciblent la membrane bactérienne, comme les peptides antimicrobiens (AMP) cationiques, constituent une source d’inspiration car ils lysent la membrane lipidique, rendant plus difficile la résistance. Afin de mieux comprendre leur mode d’action au niveau moléculaire, notre laboratoire développe des approches de résonance magnétique de l’état solide (RMN-ÉS) sur des cellules intactes. La RMN-ÉS du deutérium (2H) est un outil non destructif qui rapporte les changements de dynamique de lipides membranaires deutérés, permettant de déterminer des perturbations induites par des agents antibiotiques. L’approche développée pour deutérer la membrane lipidique d’Escherichia coli et Bacillus subtilis – représentatives des bactéries de Gram(-) et Gram(+), et de globules rouges sera présentée. Les mécanismes d’action de l’auréine 1.2 et la caérine 1.1 – des AMP excrétés par la peau de grenouilles arboricoles - déterminés par RMN-ÉS-2H in cellulo seront également montrés. Globalement, ces travaux montrent que la RMN-ÉS in cellulo est une approche prometteuse avec un grand champ d’applications.
-
Communication orale
Comment adapter les modélisations moléculaires des ARNs pour débloquer le potentiel des algorithmes d’apprentissages pour la découverte de nouvelles molécules pharmaceutiquesJerome Waldispuhl (Université McGill)
Les acides ribonucléiques (ARN) constituent une vaste classe, encore largement sous-exploitée, de cibles pharmaceutiques. À-ce-jour, nous estimons que jusqu'à 70 % de notre génome encode des ARNs, ce qui ouvre un champ immense de nouvelles possibilités. Toutefois, en raison du nombre colossal de petites molécules, le développement de méthodes informatiques capable d’identifier efficacement les candidates les plus prometteuses est essentiel. Traditionnellement, cette recherche s’appuie sur la disponibilité de structures tri-dimensionnelles de bonne qualité, mais cette information est relativement rare pour les ARNs.
Les progrès récents des technologies d'apprentissage automatique offrent de nouvelles opportunités pour repenser le pipeline de découverte de médicaments ciblant l'ARN. Dans cette présentation, je montre comment une représentation des structures d’ARNs sous la forme d’un graphe d’interactions de paires de bases non-canoniques (i.e., Au-delà des paires de bases canoniques Watson-Crick et Wobble) peut nous permettre de mieux tirer parti des algorithmes d’apprentissage pour améliorer (i) la précision des méthodes de prédiction de la structure 3D, et (ii) l'efficacité des programmes de calcul des outils d'amarrage. Cette approche a pour vocation de compléter les outils déjà disponibles et accélérer la recherche de nouveaux médicaments. -
Communication orale
Exploiter les petites molécules comme des sondes et des leads de médicament pour comprendre et cibler diverses protéinesSteven Laplante (INRS - Institut national de la recherche scientifique)
Les petites molécules peuvent être des outils précieux pour sonder les caractéristiques des protéines et elles peuvent également servir de « leads » pour découvrir de nouveaux médicaments. L'un des moyens les plus prometteurs pour découvrir ces sondes et leads se fait via la stratégie « fragment-based lead discovery » (FBLD). Le FBLD implique l’exploration des bibliothèques de petites molécules (fragments) pour identifier d'abord celles qui se lient faiblement aux protéines cibles impliquées dans les pathologies. Grâce à la chimie médicinale, ces fragments sont ensuite modifiés pour aboutir à des inhibiteurs (ou sondes) plus efficaces. Cependant, il existe des limitations majeures pour atteindre cette étape critique qui découragent de nombreux scientifiques pharmaceutiques de poursuivre cette approche. Dans cette communication, nous abordons ce problème en redéfinissant chaque étape des techniques fondamentales de la FBLD. Ainsi, de larges bibliothèques de fragments ont été développées et de nouvelles stratégies de dépistage par RMN ont été implémentées. Les propriétés des composés à l’état libre en solution ont été étudiées et des logiciels d'analyse ont été développés conduisant à un classement innovant de l'affinité par RMN. Toutes ces étapes dites «RMN pour le SAR» permettent aux chimistes médicinaux d’établir la relation de structure-activité (SAR), qui est cruciale pour rendre ces sondes et leads beaucoup plus puissantes.
Présentations éclair de la relève
Présentation de la plateforme de biologie structurale de l'Université de Montréal. Puis, quelques étudiants et étudiantes de la relève présenteront en 5 minutes le résumé de leurs recherches et profiteront du podium pour faire la promotion de leur affiche scientifique.
-
Communication orale
Plateforme de biologie structurale de l'Université de MontréalNormand Cyr (UdeM - Université de Montréal), Pascale Legault (Université de Montréal), John M. Pascal (Université de Montréal)
La plateforme de biologie structurale de l'Université de Montréal offre l’accès à plusieurs instruments scientifiques à la fine pointe de la technologie pour répondre aux questions de biologie structurale de la communauté scientifique.
Celle-ci comprend trois spectromètres de résonance magnétique (NMR) à haut champs (500 MHz, 600 MHz avec cryo-sonde et 700 MHz) contrôlés par les récentes consoles NEO de Bruker. Un instrument de BioSAXS (diffusion des rayons X aux petits angles) avec robotique pour le criblage à haut débit est aussi disponible. Cet instrument peut aisément être couplé à un système de chromatographie liquide pour les applications de SEC-SAXS.
D’autres instruments pour l’étude biophysique de molécules complémentent la plateforme de biologie structurale : un détecteur de mesure de la diffusion de la lumière sous multiples angles (SEC-MALS), un appareil de titrage calorimétrique isotherme (ITC) ainsi que plusieurs robots pour la préparation de plaques de cristallisation de biomolécules. Les utilisateurs ont aussi accès à un parc de postes informatiques pour l’analyse de données.
Du personnel qualifié est toujours disponible et s’occupera d’enseigner et d’accompagner les scientifiques dans le but de maximiser la qualité des données obtenues à la Plateforme. Un service de consultation à la recherche et un service clé en main sont aussi disponibles sur demande.
https://biochimie.umontreal.ca/plateformes-scientifiques-bmm/biologie-structurale/
-
Communication orale
Caractérisation structurale du complexe PINK1-TOM20Simon Veyron (Université McGill)
-
Communication orale
Modules structuraux d'ARN et exploration automatique de leur voisinage géométriqueVladimir Reinharz (UQAM - Université du Québec à Montréal)
-
Communication orale
Architecture moléculaire de la pointe ciliaire révélée par cryo-tomographie électroniqueThibault Legal (Université McGill)
-
Communication orale
Caractérisation des GTPases RAS dans les cellules cancéreuses à l'aide de la RMNRegina Strakhova (UdeM - Université de Montréal)
-
Communication orale
Un système génétique pour étudier les ribosomes contentant les paralogues Rps18A et Rps18B de la levure Saccharomyces cerevisiaeFrédérique Servant (UQAM - Université du Québec à Montréal)
-
Communication orale
Analyse structurale et fonctionnelle du domaine catalytique de la protéine PARP4Léonie Frigon (UdeM - Université de Montréal)
Session d’affiches
-
Communication par affiche
Analyse structurale et fonctionnelle du domaine catalytique de la protéine PARP4Léonie Frigon (UdeM - Université de Montréal)Affiche
PARP4 est une protéine qui se retrouve au niveau de l’organelle vault et qui effectue des modifications post-traductionnelles. Son rôle cellulaire précis n’est pas déterminé et il existe très peu de connaissances sur sa structure et son mécanisme de régulation. La comparaison des séquences avec d'autres PARP indique que la région catalytique de PARP4 contient un domaine hélicoïdal (HD). Le HD est un domaine autorégulateur présent dans les PARP 1, 2 et 3, qui agit pour bloquer la liaison du substrat NAD+ au site catalytique. La délétion du HD dans les PARP supprime son mécanisme d'auto-inhibition et conduit à la synthèse constitutive des modifications d’ADP-ribose. Nous avons supprimé le HD de PARP4, et observé un changement modeste de l’activité catalytique, suggérant que ce HD ne remplit pas une fonction auto-inhibitrice. Par cristallographie aux rayons X, nous avons déterminé la structure du domaine catalytique de PARP4 et confirmé la présence d'un HD ; cependant, la structure du HD est distincte de celle trouvée chez les autres PARP. Le HD de PARP4 adopte une conformation ouverte qui permet l'accès du NAD+ au site catalytique, fournissant une base structurale pour l'absence apparente d'un rôle auto-inhibiteur. Des études en cours examinent l'impact que d'autres domaines de PARP4 pourraient avoir sur la régulation de l'activité catalytique, avec l'objectif global de fournir des informations structurales et fonctionnelles qui contribuent à l'étude des fonctions de PARP4.
-
Communication par affiche
Influence de la SUMOylation sur des facteurs de transcription de plantes impliqués dans la réponse aux stress environnementauxLaurent Cappadocia (Université du Québec à Montréal), Faustine Hutinet (Université du Québec à Montréal), Laxiga Sinnathurai (UQAM - Université du Québec à Montréal)
La SUMOylation est une modification post-traductionnelle des protéines qui permet aux plantes de répondre de rapidement à des stress abiotiques comme la déshydratation ou l’exposition à des température extrêmes. La SUMOylation des protéines est typiquement dépendante de la présence de sites de SUMOylation (motif KxE où est un résidu hydrophobe, K la lysine ciblée par la SUMOylation, x un résidu quelconque et E un glutamate) sur ces protéines. Du fait de leur très petite taille, les sites de SUMOylation peuvent rapidement apparaître ou disparaître au cours de l’évolution sous l’effet de simples mutations de substitutions. Notre hypothèse est que des changements au niveau de l’abondance et de l’emplacement des sites de SUMOylation, bien qu’ils puissent contribuer à court terme à des instabilités des réseaux protéiques, contribuent à plus long terme à une adaptation rapide des plantes à de nouveaux environnements. Notre objectif est ainsi de fournir une perspective évolutive quant aux rôles de ces mutations. Plus précisément, nous déterminerons les conséquences fonctionnelles d’une augmentation ou d’une diminution du nombre de sites de SUMOylation chez la protéine HSFA2 impliquée dans la réponse aux chocs thermiques. Ceci se fera au moyen d’approches bio-informatiques, biochimiques et via la caractérisation de plantes mutantes.
-
Communication par affiche
Caractérisation structurale et biophysique des déterminants moléculaires impliqués dans l’interaction entre les protéines DELLA et SLY1 de la voie des gibbérellines chez ArabidopsiWilliam Bouard (Université du Québec à Montréal), Laurent Cappadocia (Université du Québec à Montréal), Souleimen Jmii (UQAM - Université du Québec à Montréal)
Les plantes sont des cibles directes des changements climatiques qui occasionnent un coût énergétique sur la croissance végétale. La protéine DELLA est caractérisée comme étant un répresseur de la transcription des gènes liés à la croissance. DELLA est reconnue par une Ubiquitine E3 Ligase, au niveau de la protéine SLY1 qui joue le rôle d’adaptateur spécifique de la machinerie d’Ubiquitination. L’Ubiquitination de DELLA mène à sa dégradation par le protéasome et lève la répression de croissance.
L’objectif est de caractériser les déterminants moléculaires à la base de l’interaction entre DELLA et SLY1 chez le modèle Arabidopsis thaliana.
En accord avec la littérature, nos analyses structurales des données générées par Alphafold nous ont permis d’identifier les régions impliquées dans l’interaction. Les protéines DELLA et SLY1 ont été exprimées chez E.coli et purifiées par des techniques chromatographiques. Nous avons ensuite collecté des données quantitatives de l’affinité d’interaction par la méthode de résonnance magnétique des plasmons (SPR). Un alignement de séquences de SLY1 de 151 espèces nous a permis d’identifier une semi-conservation des résidus permettant de concevoir des mutants de perte et de gain de fonction que nous avons validés par SPR. Ultimement, les connaissances obtenues serviront en agronomie pour sélectionner des plantes capables de moduler l’interaction entre DELLA et SLY1 et ainsi réguler la croissance dans des conditions environnementales défavorables. -
Communication par affiche
Architecture moléculaire de la pointe ciliaire révélée par cryo-tomographie électroniqueCorbin Black (Université McGill), Huy Bui (Université McGill), Jacek Gaertig (Université McGill), Thibault Legal (Université McGill), Max Tong (Université McGill), Melissa Valente-Paterno (Université McGill)
Les cils cellulaires sont des organites essentiels qui sont des extensions du cytoplasme. Les cils sont constitués d'une structure à base de microtubules appelée axonème. Cette circulaire est faite de 9 doublets de microtubules à la périphérie et d’une paire centrale de microtubules. Dans la plupart des types de cils, l’extrémité distale est distincte du reste du cil. Nous avons utilisé la cryo-tomographie électronique et moyenné des sous-tomogrammes pour obtenir la structure de l'extrémité ciliaire du cilié Tetrahymena thermophila. Nous montrons que les microtubules de la pointe ciliaire sont fortement liés entre eux et stabilisés par des protéines luminales ainsi que des protéines au niveau de leurs extrémités positives. Dans la région de la pointe, la paire centrale de microtubules est dépourvue des projections que l’on retrouve dans le reste du cil et tourne de manière significative. En analysant des cellules dépourvues de la protéine CEP104/FAP256 qui se trouve à la pointe ciliaire par cryo-tomographie électronique et par analyse protéomique, nous avons découvert des candidats pour le complexe de maintien de la paire centrale et expliqué les fonctions potentielles de CEP104/FAP256. Ces données apportent de nouvelles connaissances sur la fonction de la pointe ciliaire et renseignent sur les mécanismes d'assemblage et de régulation de la longueur des cils.
-
Communication par affiche
Un système génétique pour étudier les ribosomes contentant les paralogues Rps18A et Rps18B de la levure Saccharomyces cerevisiaeFrançois Dragon (Université du Québec à Montréal), Frédérique Servant (UQAM - Université du Québec à Montréal)Affiche
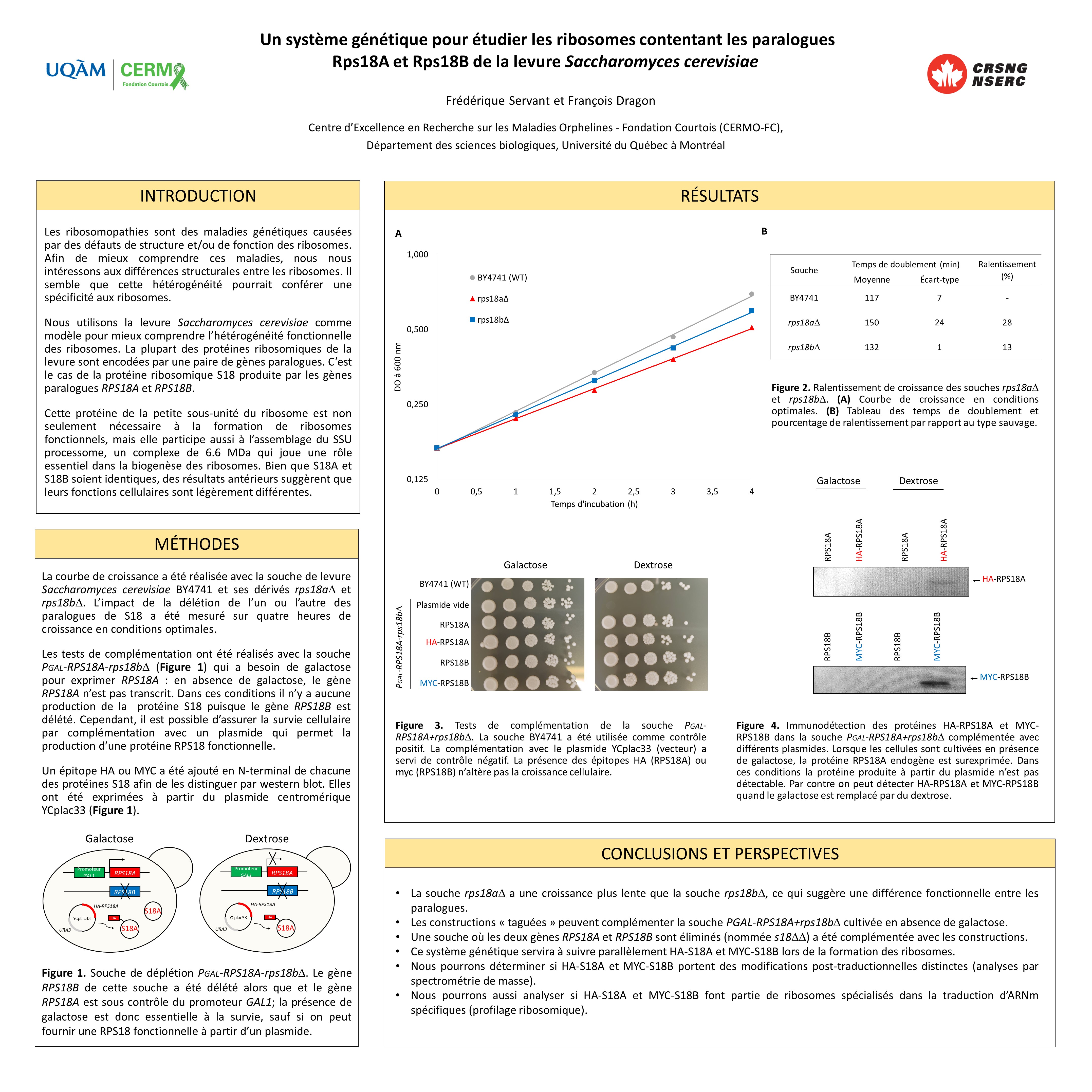
Les ribosomes d’une cellule ne sont pas tous identiques. Ils diffèrent, entre autres, par leur composition en protéines ribosomiques. Il semble que cette hétérogénéité pourrait conférer une spécificité aux ribosomes. Nous utilisons la levure Saccharomyces cerevisiae comme modèle pour mieux comprendre l’hétérogénéité fonctionnelle des ribosomes. La plupart des protéines ribosomique de la levure sont encodées par une paire de gènes paralogues. C’est le cas de la protéine ribosomique uS13 issue des gènes RPS18A et RPS18B. Afin d’étudier les différences fonctionnelles entre ces paralogues, nous avons d’abord comparé des souches délétées pour l’un ou l’autre de ces gènes. La souche rps18a∆ avait une croissance plus lente que la souche rps18b∆, ce qui suggère une différence fonctionnelle entre les paralogues. Nous avons introduit un épitope HA ou MYC à l’extrémité N-terminale des protéines uS13 afin de les distinguer et nous avons démontré que les deux constructions pouvaient complémenter une souche où les deux gènes RPS18A et RPS18B sont éliminés (nommée s18∆∆). Avec ce système génétique, nous serons en mesure de suivre parallèlement HA-S18A et MYC-S18B lors de la formation des ribosomes. Nous pourrons aussi déterminer si elles portent des modifications post-traductionnelles distinctes (analyses par spectrométrie de masse) et analyser si elles font partie de ribosomes spécialisés dans la traduction d’ARNm spécifiques (profilage ribosomique).
-
Communication par affiche
Le rôle du complexe TOM dans la stabilisation de PINK1 sur les mitochondries endommagéesSabrina Romanelli (Université McGill), Jean-François Trempe (Université McGill)
En 2031, environ 163,000 Canadiens vivront avec la maladie de Parkinson (MP), une maladie neurodégénérative accompagnées de troubles du mouvement. Les traitements existants ne traitent que les symptômes et ont des effets secondaires néfastes. Pour créer de meilleurs outils thérapeutiques, nous devons mieux comprendre les mutations qui causent la MP. Les mutations dans les protéines Parkin et PINK1 provoquent une forme autosomique récessive de la MP. Ces protéines coopèrent pour éliminer les mitochondries endommagées. Lorsque les mitochondries se dépolarisent ou accumulent des protéines mal repliées, PINK1, une kinase, s'accumule sélectivement sur les mitochondries endommagées. Elle émet un signal qui recrute Parkin, une ubiquitine ligase, pour éliminer cette portion de l’organelle. Lorsqu'il y a des dommages, PINK1 se lie au complexe TOM (translocase du complexe membranaire externe) mais leur méchanisme d'interaction est encore inconnu au niveau structural. Notre objectif est donc de mieux comprendre le rôle du complexe TOM dans la stabilisation de PINK1 sur les mitochondries endommagées. Nous utiliserons le photocrosslinking codé génétiquement et des gels natifs pour mieux comprendre les interactions entre PINK1 et le complexe TOM et déterminer la structure du complexe. Le complexe PINK1-TOM pourrait être une nouvelle cible thérapeutique pour la MP, car il s'agit de l'une des premières étapes de la cascade d'élimination de mitochondries.
-
Communication par affiche
Caractérisation structurale du complexe PINK1-TOM20Andrew Bayne (Université McGill), Jean-François Trempe (Université McGill), Simon Veyron (Université McGill)
PINK1 est une protéine kinase connue comme étant impliquée dans de nombreuses formes de la maladie de Parkinson, une maladie neurodégénérative caractérisée par des symptômes moteurs. Cette protéine est adressée à la membrane externe des mitochondries où elle sera dégradée après avoir lié la Translocase de la Membrane Externe (complexe TOM). En cas de stress mitochondrial, PINK1 ne sera plus dégradée mais accumulée à la surface de l’organelle et cette accumulation entraînera une cascade de recyclage de la mitochondrie, la mitophagie. PINK1 agit donc comme un senseur du stress mitochondrial au sein de la cellule, s’accumulant sélectivement sur les organelles endommagées. Le mécanisme de liaison de PINK1 au complexe TOM est encore inconnu mais la protéine TOM20 a récemment été identifiée comme nécessaire à son recrutement.
Dans cette étude, nous avons mené des expériences de RMN avec la protéine marquée à l’isotope 15N afin d’élucider le mécanisme moléculaire de l’interaction entre PINK1 et la portion cytoplasmique de TOM20. Avec des peptides représentant les séquences d’adressage à la mitochondrie de PINK1 (MTS 1 et 2) et de l’insert 1, une boucle flottante de la forme humaine de PINK1, nous démontrons l’interaction entre PINK1 et TOM20. De plus, le titrage de ces peptides montre une différence d’affinité de TOM20 pour ces trois différentes séquences de PINK1, élucidant le mécanisme d’interaction structurale entre ces deux protéines. -
Communication par affiche
Petites molecules inhibitrices de la kinase PINK1Nathalie Croteau (Université McGill), Shafqat Rasool (Université McGill), Tara Shomali (Université McGill), Jean-François Trempe (Université McGill), Luc Truong (Université McGill), Simon Veyron (Université McGill)
La maladie de Parkinson (MP) est une maladie neurodégénérative qui affecte de plus en plus notre population vieillissante. La forme juvénile autosomique récessive de la MP est causée par des mutations de perte de fonction dans des protéines, dont la kinase PINK1. Sous des conditions physiologiques, PINK1 est responsable de la détection des lésions mitochondriales et du déclenchement des voies de renouvellement des mitochondries. La modulation de PINK1 par de petites molécules pourrait aider à rétablir la fonction de la kinase et constituer un outil de recherche important. Notre objectif est de caractériser des petites molécules inhibitrices de PINK1 comme points de départ pour le développement de composés chimiques plus puissants et sélectifs. Les CI50 de plusieurs inhibiteurs de PINK1 ont été déterminées envers l’isoforme de PINK1 dérivé de l’insecte Tribolium castaneum (TcPINK1). Après avoir déterminé les CI50, l’activité de ces inhibiteurs devait être mesurée contre l’isoforme Homo sapiens (HsPINK1). Cependant, HsPINK1 ne peut pas être purifié à partir de bactérie donc une construction TcPINK1 humanisée a été développé dans laquelle des mutations ont été introduites dans le domaine de liaison à l'ATP, où les inhibiteurs se lient. Nous montrons que ce domaine pourrait différer entre l’espèce humaine et insecte, car la valeur des CI50 diffères. La construction humanisée de TcPINK1 servira de modèle pour le développement de dérivés de PRT062607 plus puissants envers PINK1.
-
Communication par affiche
Modules structuraux d'ARN et exploration automatique de leur voisinage géométriqueThéo Boury (Université du Québec à Montréal), Vladimir Reinharz (UQAM - Université du Québec à Montréal)
Les molécules d'acide ribonucléique (ARN) sont présentes dans toutes les cellules du vivant, et y remplissent de nombreux rôles essentiels grâce à leur capacité de se replier en diverses structures complexes. Alors que ces structures leur donnent des fonctions diverses certains blocs--- modules structuraux ---sont partagés et utilisés dans différent contexte avec parfois une grande variété de séquences. Plusieurs méthodes permettent de répertorier automatiquement ces modules identiques entre différentes molécules d'ARN. En particulier, de très bons résultats ont été obtenus en abstrayant la structure 3D atomique en un graphe, qui est annoté avec les interactions entre nucléotides et leurs 12 géométries de type Leontis-Westhof.
Nous présentons ici une méthode permettant d'échantillonner dans un ARN tous les variants d'un module structurel. La probabilité d'échantillonner dépend des différences avec le module. Trois catégories d'erreurs sont permises dans les variants (1) un mésappariement entre deux interactions, pénalisé par la différence géométrique (2) une interaction qui manque (3) une insertion de nucléotides dans le module. Une implémentation efficace dans le cadriciel ``infrared'' nous permet d'exemplifier sur le module du kink-turn notre méthode. Alors que la base de donnée RNA 3D Motif Atlas contient 18 familles de kink-turn, nous pouvons retrouver toutes les instances avec une base de quelques modules représentatifs. -
Communication par affiche
Caractérisation des GTPases RAS dans les cellules cancéreuses à l'aide de la RMNRyan C. Killoran (IRIC, Université de Montréal), Matthew J. Smith (IRIC, Université de Montréal), Regina Strakhova (UdeM - Université de Montréal)
Les petites GTPases RAS impliquées dans la signalisation intracellulaire régissent la prolifération et peuvent induire la mort cellulaire. La liaison du GTP leur permet de s’activer, changer de conformation et d’interagir avec différents effecteurs moléculaires, cela détermine alors la voie de signalisation à engager. Les mutations oncogéniques de RAS résultent en leur activation constitutive, ce qui permet aux cellules de devenir cancéreuses. Les analyses biophysiques d’activité de RAS et de leurs interactions avec d’autres protéines conduites jusqu’au maintenant étaient réalisées avec les protéines purifiées dans des conditions minimales. Cela représente une limitation importante quand nous considérons la signalisation RAS et l’activation des voies moléculaires dans le contexte d’organisation complexe des cellules vivantes. Le projet actuel porte sur l’utilisation de la spectrométrie à Résonance Magnétique Nucléaire (RMN) dans les cellules vivantes pour étudier les GTPases RAS oncogènes et leurs effecteurs en temps réel. Nos résultats indiquent qu’il possible de détecter les protéines dans les cellules vivantes, aussi bien avec le marquage uniforme au N15 qu’avec le marquage spécifique des isoleucines au C13. Les protéines transduites sont stables dans les cellules au cours du temps. Cette approche nous permettra de comprendre comment les GTPases s’activent, comment les mutations perturbent l’inactivation et ce qui détermine le choix de l’effecteur moléculaire.
-
Communication par affiche
Sondage structural SHAPE des précurseurs de miRNA let-7Pierre Dagenais (Université de Montréal), Pascale Legault (Université de Montréal), Hebatallah Samy Saad
Les miARN de la famille let-7 sont des acteurs importants dans la cellule car ils régulent la différenciation cellulaire, le développement neurologique et la suppression tumorale. La famille let-7 suit la voie canonique de biogenèse des miARN, dans laquelle le miRNA mature est produit suite au traitement de formes immatures par les ribonucléases Drosha et Dicer. Fait intéressant, dans certains types de cancer, les formes précurseurs des miARN let-7 sont présentes, tandis que les formes matures sont régulées à la baisse, ce qui suggère une mauvaise régulation de la maturation des ARN précurseurs de let-7 (pré-let-7) à l'étape de clivage par Dicer. Bien que les ARN pré-let-7 jouent un rôle important dans les cellules, leurs structures n'ont pas été étudiées en profondeur. Ainsi, il n’est pas très clair comment les caractéristiques structurales de ces pré-miARN affectent le clivage par Dicer et la liaison de protéines régulatrices. Nous avons utilisé la méthode SHAPE pour étudier la structure secondaire des ARN pré-let-7. Les résultats indiquent que les précurseurs de miRNA let-7 forment un ensemble dynamique de conformations en solution avec des caractéristiques uniques pour chaque pré-let-7. Cette diversité structurale des précurseurs de la famille let-7 contribue à l’idée que l'enzyme Dicer présente une promiscuité de substrat vis-à-vis ceux-ci.
-
Communication par affiche
Une nouvelle approche combinatoire RMN-SAXS permet de résoudre la structure du ribozyme VS en solutionPierre Dagenais, Pascale Legault (Université de Montréal)
Une stratégie couramment utilisée en sciences afin de résoudre un problème complexe consiste à le décomposer en plusieurs parties plus simples, qui peuvent alors être résolues plus facilement. Bien que les molécules d'ARN soient de nature modulaire, l'application de ce type de stratégies pour la détermination de structures tridimensionnelles d'ARN a été plutôt limitée jusqu’à maintenant. Par conséquent, nous avons développé une nouvelle approche combinatoire, que nous avons appliquée afin de déterminer des ensembles structuraux à haute résolution d’un ribozyme VS de Neurospora. En premier lieu, des études de RMN et de SAXS ont été menées sur plusieurs sous-domaines isolés qui définissent l'architecture globale du ribozyme. Ensuite, ces structures de sous-domaines ont été assemblées pour générer un large éventail conformationnel de ribozymes complets. Les meilleurs modèles structuraux ont finalement été sélectionnés en fonction de leur accord aux données expérimentales de SAXS. L'ensemble structural RMN-SAXS résultant partage plusieurs caractéristiques importantes avec les structures cristallines rapportées du ribozyme VS. Toutefois, nos modèles structuraux révèlent une différence locale située dans la tige III, qui affecte le positionnement relatif de sous-domaines du ribozyme. La découverte de ce changement conformationnel, probablement associé à la liaison du substrat, met en évidence l’utilité des approches combinatoires pour les études structurales d’ARN dynamiques.
{kind=link}
{kind=link}
Session d’affiches et cocktail
Présentations par affiche et cocktail
Mot de clôture et remise des prix
Mot de clôture et remise des prix