Enseigner des principes chimiques à l’ordinateur : une méthode efficace pour améliorer l’optimisation de géométries et de calculs d’énergies de petites molécules actives
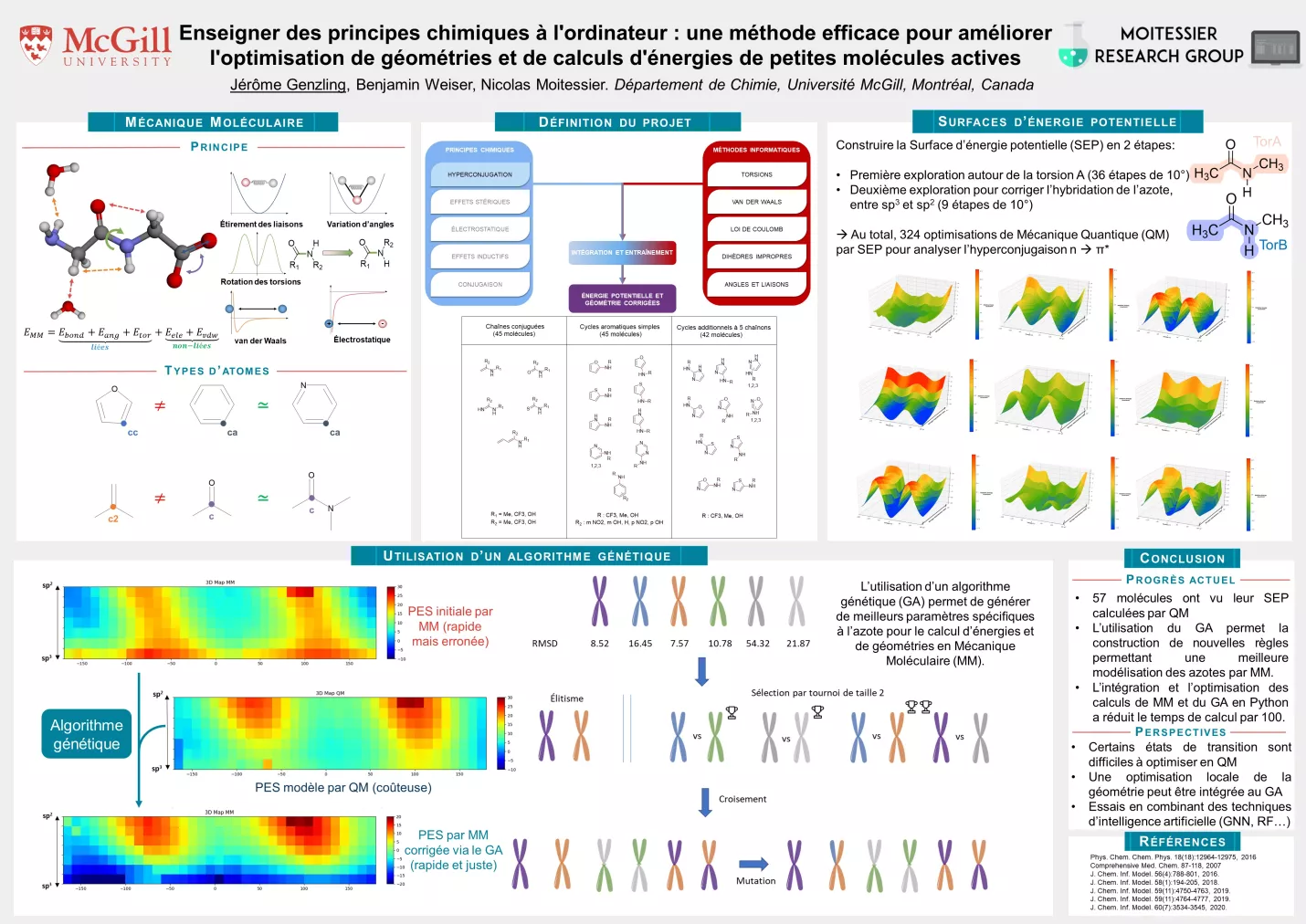
La découverte de nouvelles molécules thérapeutiques est une tâche longue et laborieuse. Afin d’accélérer ce processus, des méthodes informatiques dites in silico ont été développées. Malheureusement, ces outils sont souvent sujets à un compromis entre efficacité et temps de calcul. Notre recherche se concentre alors sur le développement d’un modèle à la fois efficace et rapide pour obtenir l’énergie potentielle et la géométrie optimisée de petites molécules d’intérêt. En effet, ces données sont notamment utiles pour améliorer des programmes de prédiction de chimie médicinale comme le criblage virtuel ou l’amarrage moléculaire.
Pour cela, nous avons développé notre propre modèle de mécanique moléculaire afin d’intégrer directement des principes connus en chimie organique tels que l’hyperconjugaison ou l’électronégativité. En effet, ces notions sont régulièrement utilisées qualitativement pour la prédiction de réactivité dans de nombreuses réflexions de chimistes organiciens. Notre modèle utilise alors ces principes pour prédire quantitativement les barrières énergétiques séparant les différentes conformations des molécules et peut être utilisé directement pour optimiser la géométrie de petites molécules d’intérêts. Cette méthodologie, dénommée H-TEQ (pour Hyperconjugaison pour la Torsion, l’Énergie et sa Quantification) a été testée sur de nombreux jeux de molécules différents de taille et de complexité graduelles et a démontré une meilleure précision que les méthodes habituelles.
Connexion requise
Pour ajouter un commentaire, vous devez être connecté.